
A fresh consideration of an enduring debate in diabetes
An international team led by Dr. Søs Skovsø, a recent post-doctoral fellow in the lab of LSI Deputy Director Dr. James Johnson, showed a causal connection between dysfunctional beta-cells and glucose-stimulated hyperinsulinemia, both of which are critical features of obesity and early type 2 diabetes.
The team addressed the mechanistic role of insulin signaling in beta-cells, a challenging area entrenched in the literature for some 20 years. Read the study
“What we investigated was the concept of pancreatic beta-cells becoming resistant,” says Skovsø. “We did that genetically, by removing the insulin receptor from beta-cells, so they could no longer respond to insulin. Beta-cells secrete insulin in response to rising blood sugar levels. Insulin then allows glucose, sugar, to enter a variety of different cells throughout the body. The big question is, upon insulin release, whether insulin has a positive or negative feedback loop on its own secretion. This has been a matter of debate for a couple of decades. Our findings showed insulin to have a negative feedback effect on beta cells. This means insulin seems to be stopping its own secretion eventually, when blood glucose levels are back to normal. We determined that stimulated by glucose, insulin-secreting beta cells were hyper-secreting insulin.”
“Doing this paper was really a dream come true,” says senior author Johnson. “There are several reasons why its important: Obviously, insulin is a critical hormone in both type 1 and type 2 diabetes. In type 2 diabetes, several tissues can be insulin resistant, meaning insulin does not work as well in these tissues. In the earliest stages of type 2 diabetes, in addition to the presence of insulin resistance, obesity and hyperinsulinemia are also often present. This means there is actually more insulin circulating in the blood. Whether hyperinsulinemia or insulin resistance and obesity comes first, and what causes what, has been debated for much longer than I have been in the field.”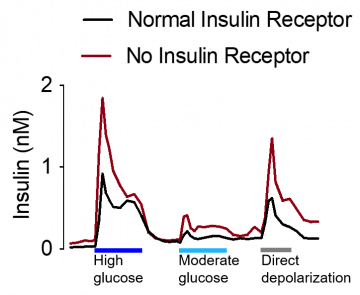
Over-secretion a bi-directional process?
Johnson has come to believe that causation is actually bi-directional in this system. “We know that insulin acts all over the body through the insulin receptors. We have known for a long time that pancreatic beta-cells that make insulin actually have insulin receptors as well. And it is an interesting observation in endocrinology that almost every hormone-releasing cell type has its own receptors on it. That is called ultra-short loop feedback and it makes sure that hormone release does not spin out of control. If there is too much insulin in the blood, it drives sugar below a safe level, and you can pass out – you can go into a coma – you can even die. Most of the hormone loops are negative, which ensures hormone secretion to stop, once enough hormone has been secreted.”
In the early days, researchers used the release of a surrogate marker for insulin, C peptide, to test what insulin does to its own release by injecting insulin into humans, as well as into large animal models. They found that insulin inhibited its own release, like other hormones, suggesting that insulin has a negative feedback effect on its own secretion to ensure hormone release doesn’t go out of control.
Those findings were challenged in 1999, by a group out of Harvard’s Joslin Diabetes Center. This was in the early days of deleting genes from specific cell types, and the researchers, led by Dr. Ronald Kahn, one of the most famous people in diabetes and insulin research, was using the best tools available at the time. The team deleted the insulin receptor gene from pancreatic beta cells. They reported that these knockout mice developed diabetes, because of a lack of insulin, inferring that insulin could no longer act through the insulin receptor to stimulate more insulin release.
“If you think about the logic of that,” says Johnson, “if these knockouts caused less insulin release, that means that the insulin receptor would normally be promoting insulin release, a positive feedback loop. But it was also an odd finding, to a lot of people, because you would have an out-of-control insulin release. How would it ever stop?”
Challenging a long established, if puzzling finding
“So, here was this result that was out in the literature for a while, and there were some other results around that time that did support those findings. About 10 years later, it became clear that most of the mouse models, the tools they were using to try to knock this insulin receptor out of the beta cells were not as specific as they were originally thought to be. They were deleting it from the brain and other cell types, which casts some doubt on that story.”
In the two decades since that time, many labs, including Johnson’s, have narrowed in on specific targeting of beta cells, helped along by the development of the right tools.
“Along comes this brilliant and ambitious postdoctoral fellow, Dr. Søs Skovsø,” adds Johnson. “I told her, ‘if we’re going to work on a project that was essentially scooped 20 years ago, we’d better get it right.’” Favours and friends were called in from near and far, creating a roster of 34 investigators, in addition to Skovsø and Johnson.
Initially, the researchers weren’t sure what they were looking for. “We were all thinking, “well, this work was done 20 years ago, and from a physiological perspective that didn’t make sense to us,” reflects Skovsø. A lot of other scientists at conferences we spoke to agreed that the biology, the physiology of positive feedback, just didn’t make sense. We wanted to find out if we would see the same thing with a new model and a new tool (a truly beta-cell specific Cre Recombinase). But we didn’t we didn’t know that the phenotype – our results – would be so small. The cool thing is, we’ve shown it in so many different ways, across different laboratories at different universities. All of the results spoke to the same thing.”
“We wanted to make sure that we were extremely thorough,” said Johnson. “We had an enormous number of mice and fed them three different diets and did measurements at four or five different ages, because we had a suspicion that the effect would be subtle. And it was.”
A subtle finding depends on inclusion
Unlike many groups that have just studied effects in males, Johnson’s group studied females as well. One of the team members was Dr. Elizabeth Rideout, CIHR Sex and Gender Science Chair in Genetics, who champions this inclusive approach in research. “As it turns out, we were lucky,” reflects Johnson. “The effect in mice is primarily in females.”
The story began to come together. “We never really saw anything like the diabetes that was reported in the Harvard paper,” continues Johnson. “There were times when we thought we were going to have to write a paper that says insulin receptors on beta cells don’t do anything. It didn’t add up, so we took a second look. I know of at least three other labs who were quietly doing the same types of experiments.”
One of those labs was that run by a world expert in insulin receptor signaling, Prof. Ernesto Bernal Mizrachi, who Johnson had trained alongside as a fellow himself. The two combined forces which in effect produced a replication study done with a slightly different model, yet still specific to beta cells, that is included as Figure 9 in the Skovsø-Johnson et al paper.
The publication was submitted for the first time in 2019. There were editors and reviewers who said the story was too complicated, and raised too many unanswered questions. The paper, questions and complicated parts intact, eventually found a good home in Nature Communications. The co-authors decided to leave them in, in hopes other researches can come at the complex biology in a different way.
Implications of the findings for humans
“In humans and animals, we’re often used to thinking of insulin resistance as one thing, but it’s becoming more and more clear that in every major cell type, the insulin resistance, the effectiveness of insulin means different things. In skeletal muscle vs the liver, versus the adipose tissue versus the brain…but people don’t think about the pancreatic beta cell. And now, we’ve added to that equation where we try to understand the earliest phases of the disease, and the pathogenesis of diabetes, and understanding the order of events, and then the initial causes. Because, then we can intervene, interrupt those initial communications between tissues.
“There all sorts of things we don’t understand, because we paint insulin resistance with one big brush,” says Johnson.
It’s vital to refine those brush strokes, because diabetes now affects at least 10 percent of the world’s population – 400 million people are living with it, and twice that many have pre-diabetes. Several billion people are at risk, by virtue of being overweight or living with obesity, according to Johnson.
“This is those very early stages where the obesity, the metabolic syndrome, the insulin resistance, this hyperinsulinemia my lab and other studies have shown at least basal hyperinsulinemia plays significant negative roles,” concludes Johnson, “so any understanding of how that arises…this might actually be a different kind of hyperinsulinemia, because it’s mostly glucose-stimulated hypersecretion. This needs to be more refined, so we can categorize these things, that’s whether where it comes down to human health, is trying to understand what causes diabetes and obesity. “
Skovsø, S., Panzhinskiy, E., Kolic, J., Cen, H. H., Dionne, D. A., Dai, X. Q., Sharma, R. B., Elghazi, L., Ellis, C. E., Faulkner, K., Marcil, S., Overby, P., Noursadeghi, N., Hutchinson, D., Hu, X., Li, H., Modi, H., Wildi, J. S., Botezelli, J. D., Noh, H. L., … Johnson, J. D. (2022). Beta-cell specific Insr deletion promotes insulin hypersecretion and improves glucose tolerance prior to global insulin resistance. Nature communications, 13(1), 735. https://doi.org/10.1038/s41467-022-28039-8